Cromatografia/Spettrometria di massa e accoppiamento GC-MS
La spettrometria di massa è una tecnica analitica che studia gli ioni in fase gas e che consente di identificare i componenti presenti all'interno di una miscela, determinarne il peso molecolare ed eseguire analisi quantitative. È una tecnica che presenta un'ampia applicabilità grazie alle sue caratteristiche intrinseche: può essere usata su campioni sia organici che inorganici e consente di eseguire analisi sia qualitative che quantitative,[1] per questo motivo le aree di interesse sono estremamente vaste e vanno dalla farmacologia alle scienze ambientali alla biologia e molte altre ancora.
Caratteristiche generali
modificaIl principio che è alla base del funzionamento dello spettrometro di massa è la separazione di ioni in fase gas in base al loro rapporto massa/carica, che vengono poi inviati ad un rivelatore che converte l'abbondanza dei singoli ioni in un segnale elettrico.[2]
Per quanto concerne i limiti di rivelabilità associati a questa tecnica sono molto bassi e spesso inferiori a 1 ppb nel caso della spettrometria di massa atomica,[3] è poi una tecnica caratterizzata da un ampio range di applicabilità in quanto è possibile determinare ioni aventi peso molecolare fino a 108 Da.[4]
Un grande limite legato a questa tecnica è che non è in grado di separare composti diversi, per questo motivo viene infatti accoppiata ad una tecnica separativa come la cromatografia liquida o la gascromatografia. Ovviamente, essendo differenti le condizioni operative richieste da queste due tecniche bisognerà utilizzare un adeguato sistema di interfaccia che consenta ai due strumenti di lavorare insieme.
La strumentazione che viene impiegata per la spettrometria di massa può essere schematizzata nel seguente modo:

Sistema di introduzione del campione
modificaIl campione può essere introdotto sotto diverse forme, potendo trovarsi allo stato solido, liquido o gas; per questo motivo questa tecnica si presta bene ad essere accoppiata sia alla cromatografia liquida che alla gascromatografia. Aspetto critico è quello di creare un'interfaccia adeguata che consenta di mantenere l'alto vuoto all'interno della strumentazione: lo spettrometro di massa infatti lavora ad alto vuoto in modo tale da minimizzare gli urti tra gli ioni di analita generati e altre particelle in fase gas presenti. Se così non fosse gli ioni generati non raggiungerebbero il rivelatore a causa dell'elevata temperatura presente all'interno dello strumento e della reattività intrinseca degli ioni. Per queste ragioni il sistema di introduzione del campione è quindi il punto critico dell'accoppiamento delle due tecniche e deve garantire la corretta introduzione del campione senza compromettere il vuoto del sistema.
Sorgente ionica
modificaÈ il sistema all'interno del quale avviene la ionizzazione degli ioni. A seconda del metodo utilizzato per la loro generazione si avranno due diversi tipologie di sorgenti:
- Ionizzazione hard - la quantità di energia che viene fornita alle molecole di analita è molto elevata. La ionizzazione utilizzata in questo caso è detta ionizzazione elettronica e si ottiene bombardando l'analita con un fascio di elettroni.
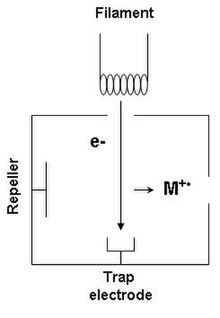
Sorgente a ionizzazione elettronica - Ionizzazione soft - la quantità di energia che viene fornita alle molecole di analita è sufficiente alla sola ionizzazione.

La sorgente a ionizzazione elettronica (EI - Electron Ionization), può essere applicata solamente a campioni in fase gas, per questo motivo viene solitamente usata nel caso si abbia a che fare con analiti sufficientemente volatili. La sorgente è costituita da un filamento di tungsteno o rodio il quale emette elettroni perpendicolarmente rispetto alla direzione di avanzamento del campione. Il fascio di energia proveniente dal filamento è solitamente di 70 eV che è una quantità molto più elevata di quella necessaria per la sola ionizzazione del campione. Quando le molecole di analita incontrano il fascio di elettroni ad alta energia si ionizzano a seguito degli urti con gli elettroni e si generano quindi ioni positivi. Gli ioni positivi così generati vengono quindi accelerati da un sistema di lenti a potenziale crescente: in questo modo i cationi vengono spinti verso l'analizzatore mentre i pochi ioni negativi che si sono generati vengono accelerati verso il repeller. Tutte le specie in uscita dalla sorgente hanno energia cinetica per via dell'accelerazione delle lenti a potenziale crescente.
La quantità di anioni che viene generata con questa tipologia di sorgente è molto piccola: la loro formazione è infatti sfavorita e porta ad un rapporto ioni positivi/negativi di circa 10.000/1. Per quanto riguarda invece il rapporto tra il numero di molecole inserite e in numero di molecole ionizzate utilizzando un fascio di elettroni di 70 eV questo è di circa 1/1000.
A causa dell'elevata energia del fascio di elettroni che viene utilizzato, oltre alla ionizzazione si ha anche la frammentazione degli analiti in specie più piccole e il cui schema di frammentazione dipende dalla natura chimica della specie così come la sua tendenza a frammentarsi. Ogni evento di frammentazione da luogo alla formazione di due o più specie chimiche di cui una sola sarà carica, le altre invece saranno neutre: la carica elettrica totale si conserva, per cui solamente la specie carica sarà rivelabile dallo strumento, le specie neutre infatti non raggiungeranno neanche il rivelatore. Frammenti identici formati da percorsi di frammentazione differenti daranno un unico segnale la cui intensità sarà data dalla somma delle intensità dei singoli ioni.
Si lavora con un fascio di elettroni da 70 eV perché se si operasse ad energie maggiori si avrebbe una frammentazione eccessiva dell'analita e sarebbe quindi molto più complicato risalire alla struttura della sostanza da cui si è generato. Ad energie inferiori invece si avrebbe una ionizzazione molto ridotta che porterebbe alla generazione di un numero troppo piccolo di ioni, il che sarebbe poco funzionale per l'analisi qualitativa: 70 eV è quindi un valore che costituisce un buon compromesso tra questi due effetti.
Analizzatore
modificaL'analizzatore di massa è un dispositivo che è in grado di distinguere gli ioni in funzione del loro rapporto massa su carica (m/z).
La capacità di uno spettrometro di massa di distinguere tra masse è definita in termini di risoluzione:[5]
- con m → massa nominale del primo picco
- Δm → differenza tra le masse dei due picchi adiacenti.

A seconda del tipo di impiego la risoluzione richiesta ad uno spettrometro di massa può anche essere molto diversa. nel caso in cui si abbia a che fare con ioni aventi la stessa massa nominale è necessario avere un analizzatore con una risoluzione molto alta, di diverse migliaia. Qualora invece gli ioni da discriminare abbiano masse molecolari che differiscono le une dalle altre per almeno un'unità di massa, la risoluzione richiesta dallo strumento sarà ovviamente minore rispetto al caso precedente.[6]
A seconda del principio alla base del funzionamento possiamo distinguere i seguenti tipi di analizzatore:
- a settore magnetico;
- quadrupolare;
- a tempo di volo.
L'analizzatore a settore magnetico è il primo ad essere stato inventato, è molto costoso e voluminoso. È costituito da un magnete curvo e il suo funzionamento si basa sulla deviazione che ogni particella carica in movimento subisce dalla sua traiettoria rettilinea in seguito all'applicazione di un campo magnetico ad essa perpendicolare.
La deviazione nella traiettoria che lo ione subisce è espressa dalla seguente equazione:[7]
- con r → raggio di curvatura del magnete;
- B → campo magnetico;
- vs → potenziale di accelerazione.

Dalla seguente formula si evince che il raggio di curvatura del magnete che lo ione deve attraversare nel suo tragitto influisce in modo direttamente proporzionale al rapporto m/z selezionato. Mantenendo costante il potenziale di accelerazione l’unico fattore che incide sulla selezione del rapporto m/z è il campo elettrico applicato, per questo motivo variando lentamente nel tempo il valore che questo assume, si riesce ad ottenere la separazione degli ioni.

L’analizzatore quadrupolare è formato da quattro elettrodi a sezione circolare o iperbolica a due dei quali viene fornita carica positiva, mentre ai restanti due carica negativa. I due elettrodi aventi stessa carica sono messi agli antipodi e collegati tra loro affinché il potenziale applicato sia identico. Ad una coppia di elettrodi viene poi applicato un potenziale continuo mentre all’altra si applica un potenziale alternato oscillante a radiofrequenze.
Il potenziale totale generato dal quadrupolo, ovvero il campo che va ad agire sugli ioni, è in definitiva un campo oscillante che viene descritto dalla seguente espressione:

con Ω = frequenza angolare del campo alternato (rad s-1)
Per eseguire l’analisi si va ad incrementare il valore di tensione continua e alternata applicata in modo tale che il rapporto Vcost / Valt rimanga costante. Con l’opportuna regolazione delle tensioni infatti, viene creato un percorso stabile solamente per ioni aventi un determinato rapporto m/z: questi saranno in grado di descrivere la traiettoria sinusoidale necessaria per arrivare fino al rivelatore, gli altri invece verranno eliminati lungo il percorso in quanto descriveranno oscillazioni di ampiezza via via crescente, per cui procedendo lungo il percorso andranno a collidere contro una delle sbarre costituenti un elettrodo. Lo spettro di massa viene quindi ottenuto scansionando le tensioni che vengono applicate agli elettrodi.
Il tempo necessario per eseguire la scansione completa è piuttosto breve, inoltre la capacità di trasmissione è elevata. Uno dei più grandi svantaggi è costituito dal fatto che la risoluzione è piuttosto bassa, il che la rende adatta ad analisi in accoppiata con cromatografia liquida o gascromatografia, ma non alla rivelazione di ioni con massa superiore ai 1000 Da.
L’analizzatore a tempo di volo (TOF - Time Of Flight) è formato da un lungo tubo percorso da ioni precedentemente accelerati da una differenza di potenziale.
Dal momento che l’energia cinetica di uno ione è definita come:
dove m è la massa dello ione e v la sua velocità
Ma allo stesso tempo dipende dal potenziale di accelerazione applicato e dalla stessa carica dello ione come descritto dalla seguente equazione:
,
dove q è carica totale
Unendo le informazioni ricavabili dalle precedenti equazioni si ricava che la velocità di uno ione che lascia la sorgente è uguale a:[8]



dove Vacc è il potenziale di accelerazione che subisce lo ione
Si consideri che il tempo necessario allo ione per coprire la distanza d che lo separa dal rivelatore è definito come:
per cui andando a sostituire il valore di velocità precedentemente ottenuto nella formula qui riportata del tempo si ricava che:


Dalla precedente formula si evince che il tempo impiegato dagli ioni per percorrere una distanza fissa verso il rivelatore è inversamente proporzionale alla massa degli ioni stessi. Per questo motivo ioni con basso valore m/z arrivano al rivelatore più velocemente di quelli con elevato valore m/z: a parità di carica saranno gli ioni più leggeri a raggiungere per primi il rivelatore.
Affinché la separazione sia utile è necessario che tutti gli ioni partano contemporaneamente dalla sorgente ionica, in questo modo tutti gli ioni aventi lo stesso rapporto m/z arriveranno contemporaneamente al rivelatore e tutti gli ioni vengono rivelati in sequenza a seconda del valore del loro rapporto massa/carica.
Gli analizzatori a tempo di volo sono in generale strumenti semplici e robusti, dotati di un intervallo di masse rivelabili praticamente illimitato e i tempi di analisi sono piuttosto brevi: il tempo di acquisizione di uno spettro di massa completo è nell’ordine dei µs.[9] Il loro più grande limite è però dato dal fatto che sono caratterizzati da risoluzione e sensibilità piuttosto basse se confrontate con quelle di altri analizzatori, questo perché il numero di ioni che viene rivelato è molto piccolo.
La risoluzione dell’analizzatore a tempo di volo si ricava facendo alcune considerazioni in merito al rapporto m/z e il tempo di volo. Innanzitutto si isola il rapporto m/z:

Da cui si ricava che:

E quindi:

Da cui si evince che la risoluzione dell’analizzatore a tempo di volo è pari a:
- con m e t rispettivamente massa e tempo di volo dello ione;
- Δt e Δm l’ampiezza a mezza altezza dei picchi;
- d la distanza percorsa dallo ione;
- Δx lo spessore di un pacchetto di ioni che arriva al rivelatore.

Dal momento che la risoluzione è proporzionale al tempo di volo e al percorso, per migliorare la risoluzione è possibile agire in due modi:
- Allungare il tubo dell’analizzatore in modo da allungare il cammino che ogni ione deve compiere per arrivare al rivelatore – in questo modo però si aumenta la perdita di ioni a causa della loro collisione con le molecole di gas;
- Abbassare la tensione di accelerazione per aumentare il tempo di volo, questo comporta però l’abbassamento della sensibilità.
È quindi necessario trovare un adeguato compromesso che consenta di avere una buona risoluzione e sensibilità, questo viene fatto utilizzando un tubo lungo 1-2 metri e usando una tensione di accelerazione di 20 kv.[10]
Rivelatore
modificaI rivelatori utilizzati nella spettrometria di massa sono strumenti in grado di generare una corrente elettrica la cui intensità è proporzionale all'abbondanza degli ioni incidenti. Ne esistono di diverse tipologie e la scelta di uno rispetto ad un altro dipende dal tipo di applicazione analitica che si svolge e dalla geometria della strumentazione.
Dal momento che il numero di ioni che superano l'analizzatore è solitamente piuttosto piccolo, è spesso necessaria un'amplificazione del segnale.
Il primo rivelatore impiegato nella spettrometria di massa è il piatto fotografico. Il suo funzionamento si basa sul fatto che gli ioni con lo stesso rapporto m/z raggiungono la piastra nello stesso punto: in questo modo è possibile rivelare simultaneamente un vasto numero di ioni con rapporto massa/carica differente. L'abbondanza relativa dei singoli ioni viene invece stimata sulla base dell'oscurità delle macchie che rimangono impresse sulla lastra fotografica. Questo metodo di rivelazione consente di avere un valore di abbondanza ovviamente approssimativo, per questo motivo è ormai in disuso.
Un'altra tipologia di rivelatore è a coppa di Faraday il quale è costituito da una coppa metallica o cilindrica con un piccolo foro la quale è collegata a terra con un resistore. Gli ioni che riescono a raggiungere l'interno del cilindro vengono neutralizzati attraverso la donazione o acquisizione di elettroni dalle pareti del rivelatore contro cui impattano: questo processo di scarica produce un passaggio di corrente che viene amplificata e rivelata. Questa tipologia di rivelatore presenta innumerevoli fonti di errore, tra queste ad esempio la presenza di elettroni secondari i quali vengono generati nel momento in cui un elettrone impatta contro la parete del rivelatore: se non vengono eliminati per tempo vengono rivelati dal rivelatore falsando il segnale. È quindi necessario adottare un adeguato sistema di cattura di elettroni oppure adottare delle accortezze per ridurne la produzione o impedirne la rivelazione. Una soluzione può essere realizzata ricoprendo le pareti del rivelatore con carbone, in quanto produce un minor numero di elettroni secondari. Anche la forma della coppa di Faraday è estremamente importante: infatti, insieme all'utilizzo di un campo magnetico debole impedisce agli elettroni secondari di uscire ed essere rivelati. Questa serie di fattori incide soprattutto sulla sensibilità (che è piuttosto ridotta) e sulla bassa velocità di risposta.
Il rivelatore più comunemente usato è il moltiplicatore di elettroni a dinodi discreti il cui principio di funzionamento è analogo a quello di un tubo fotomoltiplicatore. Gli elettroni e ioni che arrivano dall'analizzatore vengono fatti impattare contro un catodo di Cu-Be generando elettroni secondari i quali a loro volta vengono indirizzati e accelerati verso il dinodo successivo il quale viene mantenuto ad una tensione positiva sempre più elevata. Il numero di dinodi può variare da 12 fino a 20 unità.[11]

Un'altra variante di questa tipologia di rivelatore è il moltiplicatore di elettroni a dinodo continuo in cui i dinodi discreti sono sostituiti da un dinodo continuo di forma solitamente a corno, le cui pareti sono costituite da vetro drogato Pb.[12][13] La differenza di potenziale viene applicata lungo tutto il rivelatore. Gli elettroni e gli ioni entrano nel rivelatore dalla parte dell'apertura maggiore, impattano contro la superficie del tubo e generano elettroni secondari che vengono accelerati verso l'uscita del rivelatore: prima di arrivare alla fine della sua lunghezza però impatteranno più volte con le pareti amplificando il segnale per effetto cascata. L'elevato fattore di amplificazione che si può ottenere in entrambi i casi (circa 107)[14] e la velocità di risposta di questo rivelatore lo rendono estremamente adatto. L'unica problematica è legata al fatto che il fattore di rivelazione dipende fortemente dalla velocità di impatto degli ioni rivelati così come dalla loro massa e carica, per questo motivo l'efficienza di rilevamento è molto inferiore per ioni con elevato m/z e per ioni molto lenti. L'utilizzo dei dinodi discreti e l'applicazione di un potenziale via via crescente aiuta ad accelerare gli ioni cosicché si riesca ad ottenere una buona efficienza di rilevamento anche per gli ioni più lenti. In particolare il moltiplicatore a dinodi discreti viene impiegato per la rivelazione di ioni ad alta massa e a bassa energia cinetica.
Accoppiamento GC-MS
modificaL'utilizzo dello spettrometro di massa come rivelatore per la gascromatografia presenta molti vantaggi:
- è estremamente specifico nella selezione di ioni da quantificare;
- è uno dei rivelatori più sensibili in assoluto;
- è estremamente versatile.
L'idea dell'accoppiamento nasce dal fatto che queste due tecniche svolgono funzioni analitiche complementari: la gascromatografia consente la separazione di miscele complesse, mentre la spettrometria di massa consente la caratterizzazione e la quantificazione dei composti.[15]
Il problema legato all'accoppiamento di queste due tecniche è legato alle differenti condizioni operative richieste, che portano alla necessità di creare di un'interfaccia adeguata che permetta di far lavorare insieme i due strumenti: innanzitutto la colonna cromatografica lavora sotto pressione, mentre lo spettrometro di massa lavora in alto vuoto. Inoltre il flusso delle colonne cromatografiche, soprattutto quelle impaccate, è molto importante e per niente trascurabile, soprattutto in ambito di cromatografia liquida. Un altro aspetto estremamente importante nasce dalla necessità di operare una scansione molto velocemente: gli analiti devono essere rivelati non appena escono dalla colonna o il rischio è quello di vanificare la separazione, in più l'eventuale spurgo della colonna crea molti problemi.
La prima cosa da fare per creare un'interfaccia adeguata è quella di impoverire il campione proveniente dalla colonna cromatografica di fase mobile in modo da ridurre il flusso in arrivo allo spettrometro di massa, pratica particolarmente importante nel caso di colonne impaccate che utilizzano importanti volumi di fase mobile. Un'interfaccia molto usata prevedeva l'utilizzo di un tubo vetro poroso all'interno del quale veniva fatto passare il flusso in uscita dalla colonna. Il tubo di vetro veniva quindi sottoposto all'esterno all'alto vuoto, il che forzava il gas carrier, solitamente elio, ad uscire dal tubo il quale era dotato di pori di dimensioni tali da permettere solo il passaggio del gas carrier ma non dell'analita: si sfruttava infatti le ridotte dimensioni dell'elio che è il gas carrier più usato. Il problema principale era però dovuto al fatto che una quantità non trascurabile di analita veniva comunque persa (circa il 20-50%).[16] Dal momento che si è passati all'impiego di colonne capillari, questa tipologia di interfacce non è più necessaria in quanto queste colonne hanno un flusso che è già abbastanza ridotto da essere compatibile con la portata richiesta dallo spettrometro di massa. L'unica accortezza che va fatta è legata al mantenimento di una temperatura adeguata: si utilizza infatti una piccola sezione di tubo riscaldato al termine della colonna che serve ad evitare la ricondensazione delle sostanze che sono state separate con la cromatografia in modo quindi che si mantengano in fase gas al momento dell'iniezione nello spettrometro, il che causerebbe l'allargamento dei picchi o anche la cattura di sostanze altobollenti che non arriverebbero quindi allo spettrometro di massa.[17]
Come in gascromatografia c'è il rischio della degradazione termica degli analiti, motivo per il quale il controllo termico è di fondamentale importanza: è per questo che la temperatura di iniezione è solitamente non eccessivamente alta. Va inoltre tenuto a mente che gli eventuali prodotti di degradazione termica possono essere identificati durante l'analisi con uno spettrometro di massa. Per prevenire la degradazione termica degli analiti viene solitamente utilizzato l'iniettore on column.
Dal momento che la degradazione di alcune componenti può anche essere causata dalla presenza di gruppi silaniolici della superficie del vetro del liner di iniezione, una buona soluzione è costituita dall'impiego di agenti silanizzanti che convertono i gruppi silanolici in eteri trimetilsililici.[18]
Un altro aspetto che va ben tenuto in considerazione al momento dell'accoppiamento di queste due tecniche è legato alla portata del flusso che può essere sopportato dallo spettrometro di massa, il che costituisce un elemento limitante. Questo è solitamente di circa 1 mL/min, anche se può essere leggermente più alto se si usano accortezze particolari. In linea generale vengono quindi impiegate colonne dal diametro da 0,25 mm o tuttalpiù da 0,32 mm, non è però possibile usare colonne megabore in quanto hanno dimensioni eccessive.[19]
L'iniezione non rappresenta comunque un fattore limitante in quanto vanno bene sia iniettori split-splitless in entrambe le modalità, sia gli on column injector. Le considerazioni fatte nelle sezioni precedenti riguardo ai criteri di scelta si applicano allo stesso modo. Unico aspetto da controllare è che le connessioni tra i due siano a tenuta stagna per evitare perdite di pressione che possono inficiare sul corretto funzionamento dello strumento. Quando possibile è preferibile usare l'iniettore split-splitless in modalità split per evitare la contaminazione da solventi delle pompe da vuoto. L'unico caso in cui l'iniettore split-splitless in modalità split è da evitare è nel caso in cui l'analita da analizzare sia presente in tracce in quanto il rischio è quello di perderne una quantità importante e non riuscire quindi a rivelarlo. Dal momento che nelle iniezioni in modalità splitless il campione viene iniettato tal quale, per proteggere la colonna da ostruzioni o contaminazioni causate da sostanze eventualmente presenti nel flusso di analita oggetto di analisi, viene utilizzato spesso un setto rivestito di materiale adsorbente, la cui funzione è proprio quella di intrappolare tali sostanze, non volatili e polari.[20]
Un altro problema nell'accoppiamento di queste due tecniche è dato dal fatto che, mentre il rivelatore analogico tradizionale misura una proprietà del gas in arrivo ed è quindi in gradi di fornire un segnale continuo, lo spettrometro di massa esegue singole scansioni ad intervalli. Per avere un cromatogramma adeguato è quindi necessario avere un elevato numero di scansioni in un breve lasso di tempo e di avere quindi scansioni estremamente rapide: in questo modo infatti, avendo un elevato numero di punti sul grafico, è possibile approssimare il picco in modo adeguato e quantificare l'area sottesa efficacemente.
Un ultimo ma non meno importante problema è dato dal fenomeno di spurgo della colonna: mano a mano che la temperatura aumenta durante l'eluizione a gradiente di temperatura, la fase stazionaria costituente l'impaccamento della colonna tende a disgregarsi ed a lasciare frammenti che reggiungono lo spettrometro di massa e vengono quindi rivelati causando l'innalzamento della linea di base del cromatogramma. Questo fenomeno è difficile da evitare ma può essere ridotto scegliendo una fase stazionaria sufficientemente stabile.
Tutto ciò che viene inserito nella colonna cromatografica entra e viene rivelato dallo spettrometro di massa, eccezion fatta per le componenti del campione che rimangono trattenute in colonna. Per i componenti volatili non si pone alcun tipo di problema dal momento che, al termine dell'analisi, vengono poi pompati via tramite il sistema di vuoto ma, nel caso in cui si abbia a che fare con materiali semi volatili, o ancora peggio non volatili, la situazione assume un'importanza ben diversa: queste componenti possono depositarsi nella fonte di ioni dello spettrometro di massa, diminuendone la sensibilità e portando ad aver bisogno di una maggior manutenzione. Per evitare queste spiacevoli conseguenze si ricorre all'eliminazione di queste sostanze in modo da preservare lo spettrometro di massa. I composti inorganici non volatili possono essere rimossi per scambio ionico o tramite estrazione, mentre i composti organici e polari tramite gel di silice o fluorisil. Dal momento eseguire queste eliminazioni richiede la manipolazione del campione, bisogna eseguire questi metodi con molta attenzione e solo se realmente necessario in quanto il rischio è quello di avere una perdita involontaria di una parte di analita.
Anche in GC-MS si può ricorrere alla derivatizzazione delle molecole di campione con lo scopo di migliorare la simmetria del picco, la volatilità e la stabilità termica di un dato componente per migliorarne la separazione gas cromatografica o anche per consentire di avere una migliore selettività e limiti di rivelabilità nell'analisi spettrofotometrica. A tale scopo il metodo che viene comunemente utilizzato prevede la derivatizzazione di uno o più atomi di idrogeno attivi (ovvero degli idrogeni appartenenti a un gruppo -OH, -COOH, -NH2, ecc.) attraverso una serie di reattivi come ad esempio N,O-bis(trimetilsilil)acetammide oppure bis(trimetilsilil)frifluoroacetammide.[21] In questo modo tale idrogeno viene sostituito con un gruppo trimetilsilil modificando la massa del composto che verrà quindi rivelato a valori maggiori nello spettro di massa.
Altro aspetto da tenere a mente nell'accoppiamento di queste due tecniche è la scelta del solvente: deve essere compatibile con entrambe e non deve dare picchi di interferenza, per questo motivo si deve selezionare un solvente che non fornisca picchi che ricadano nell'intervallo m/z di interesse nell'analisi.
Esempio di applicazione
modificaLa metodologia analitica che prevede l’accoppiamento GC-MS è, come abbiamo visto nel corso di questo capitolo, una tecnica estremamente efficace nella separazione e determinazione di analiti in matrici anche molto complesse. In questa sezione verrà proposta la costruzione di un metodo analitico che prevede la determinazione quantitativa degli IPA in un campione di sedimento marino utilizzando la tecnica accoppiata GC-MS usando come riferimento il metodo analitico ufficiale dell’ISS.[22]
Gli idrocarburi policiclici aromatici sono prodotti derivati dalla combustione incompleta e la pirolisi di materia organica e la loro determinazione è estremamente importante in quanto 16 di loro presentano attività cancerogena (naftalene, acenaftilene, acenaftene, fluorene, fenantrene, antracene, fluorantene, pirene, benzo[a]antracene, crisene, benzo[a]fluorantene, benzo[k]fluorantene, benzo[a]pirene, indeno[1,2,3-c,d]pirene, dibenzo[a,h]antracene, benzo[g,h,i]perilene). La loro presenza viene monitorata per valutare l’impatto ambientale che i processi umani hanno e per farlo tale analisi viene eseguita su matrici diverse (acqua, sedimenti..). Per prima cosa bisogna andare a definire quelle che sono le condizioni di esercizio per entrambe le tecniche e costruire un’interfaccia che funzioni.
Per quanto riguarda la gascromatografia si sono usati i seguenti parametri:

- colonna apolare Phenomenex ZB-5 (fenilmetilsilossano al 5%), 30 m x 0,25 mm I.D. x 0,25 micrometri;
- iniettore on-column;
- volume di iniezione 0,5 µL;
- gas carrier costutuito da He a flusso costante di 1,4 mL/min;
- programmata di temperatura come in figura.
Per quanto riguarda invece i parametri usati per la spettrometria di massa abbiamo:
- energia degli elettroni impostata a 70 eV;
- temperatura del transfer line di 280 °C;
- temperatura della sorgente di 250 °C;
- temperatura del quadrupolo di 100 °C.
È poi di necessaria importanza avere a disposizione uno standard certificato contenente i 16 composti da monitorare che verranno poi analizzati nelle condizioni sopra riportate così da ottenerne i tempi di ritenzione: questo ci consentirà di identificare i vari composti presenti nel campione attraverso il semplice confronto dei tempi di ritenzione ed eseguire così l’analisi qualitativa. Per l’analisi quantitativa si esegue invece una scansione SIM (ovvero Selected Ion Monitoring - scansione che consente di monitorare solo una piccola quantità di rapporti m/z corrispondenti a ioni rappresentativi dell'analita) su ioni scelti opportunamente per ciascun analita. Prima di eseguire questa tipologia di analisi è prima necessario eseguire una calibrazione: in questo caso si propone il metodo di diluizione isotopica, un sistema di standard interno che prevede l’aggiunta al campione di una concentrazione nota di analita deuterato.
Si costruisce quindi la retta di taratura preparando più soluzioni standard a concentrazione crescente e ponendo sull’asse delle ordinate il rapporto tra le aree di analita e di standard interno e sull’asse delle ascisse il rapporto delle concentrazioni delle stesse, in questo modo è infatti possibile anche tenere conto dell’efficacia della procedura di estrazione degli analiti dalla matrice iniziale.
Ottenuta quindi la retta di taratura del tipo y = mx + q si ricava la concentrazione di analita (espressa in ppb) con la seguente formula:

A questo punto, noto il volume iniettato e nota la massa del campione analizzato si ricava la concentrazione dell’analita espressa in ng/g:

Note
modifica- ↑ Skoog, p. 802
- ↑ Skoog, p. 804
- ↑ Skoog, p. 811
- ↑ Hoffmann, p. 9
- ↑ Skoog, p. 805
- ↑ Skoog, p. 805-806
- ↑ Hoffmann, p. 144
- ↑ Hoffmann, p. 126
- ↑ Hoffmann, p. 127
- ↑ Hoffmann, p. 129
- ↑ Hoffmann, p. 177
- ↑ Skoog, p. 807
- ↑ Hoffmann, p. 178
- ↑ Skoog, p. 807
- ↑ Herbert, p. 253
- ↑ Grob, p. 346
- ↑ Grob, p. 347
- ↑ Grob, p. 347
- ↑ Grob, p. 346
- ↑ Grob, p. 346
- ↑ Grob, p. 345
- ↑ E. Menichini e G. Viviano (a cura di), Gruppo di lavoro Istituto Superiore di Sanità "Metodiche per il rilevamento delle emissioni in atmosfera da impianti industriali", Determinazione degli idrocarburi policiclici aromatici (IPA). Metodo gascromatografico, Rapporti ISTISAN 97/35, settembre 1997, ISSN 1123-3117. https://www.osti.gov/etdeweb/servlets/purl/618097